ILAE 2012 classification of electroclinical syndromes
and other epilepsies
Specific Epilepsy Syndromes – Genetic Generalised
Epilepsy
Idiopathic Generalised Epilepsies
Childhood Absence Epilepsy (CAE)
Juvenile Absence Epilepsy (JAE)
Early onset absence epilepsy (not accepted as official
category in ILAE)
Genetic Epilepsy with febrile seizures
Other Genetic Generalised Epilepsies
Epilepsy with Myoclonic Absence (EMA)
Epilepsy with eyelid myoclonia (EEM) (Jeavons syndrome)
GLUT 1 Deficiency (aetiological classification rather
than true syndrome)
Specific epilepsy syndromes – “Symptomatic”
generalised epilepsy
Epilepsy with myoclonic atonic seizures (EMAtS) (Doose syndrome)
Progressive Myoclonic Epilepsies
Progressive, dementing conditions
Familial Adult Myoclonic Epilepsy (FAME)
Concept of Developmental/Epileptic Encephalopathies
Specific epilepsy syndromes – Focal epilepsies
Sleep related hypermotor epilepsy (SHE)
Familial Mesial Temporal Lobe Epilepsy (FMTLE)
Familial Focal Epilepsy with Variable Foci (FFEVF)
Epilepsy with Auditory Features (EAF)
Self-Limited Focal Epilepsies (SeLFEs)
Childhood occipital visual epilepsy (COVE)
Photosensitive occipital lobe epilepsy (POLE)
Self-Limited Epilepsy with Centrotemporal Spikes
(SeLECTS)
Self-Limited Epilepsy with Autonomic Seizures (SeLAS)
Specific epilepsy syndromes – “Symptomatic/Lesional”
Focal Epilepsies
Mesial Temporal Lobe Epilepsy with hippocampal sc
Autosomal Dominant Sleep-related Hypermotor Epilepsy
(ADSHE)
Autosomal Dominant Epilepsy with Auditory Features
Familial Focal Epilepsy with Variable Foci (FFEVF)
Familial mesial temporal lobe epilepsy
Epilepsy Syndromes
Classification
• Individual seizures can be classified by type
o
Focal
o
Generalised
o
Unknown
• Aetiology is next step in defining the epilepsy
o Three categories:
- Genetic
- Structural/Metabolic
- Unknown
• Attempt to group seizures according to electro-clinical features into a particular group
o Electro-clinical syndromes
- Reasonably well defined, distinctive clinical disorder
o Constellations (Surgical syndromes)
- Groups of features with tend to occur together but not a clearly distinctive syndrome
Individual Seizure Types
• Focal
o Aura (Sensory)
o Motor
o Autonomic
o With loss of awareness (dyscognitive)/ With preserved awareness
o May evolve to: Bilateral convulsive seizure
• Generalised
o Tonic-clonic
o Absence
- Typical
- Atypical
- With special features:
• Myoclonic absence
• Eyelid myoclonia
o Clonic
o Atonic
o Myoclonic
- Myoclonic
- Myoclonic atonic
- Myoclonic tonic
Aetiology
• Genetic
o The result of a known or presumed genetic mutation in which seizures are the core symptom of the disorder
• Structural/Metabolic
o Structural (e.g. tumour or genetic malformation – however not classified as genetic as there is another step interposed between the gene mutation and the epilepsy)
o
• Unknown
Classification
- old
- Partial
/Localisation-related/ Focal seizures
- Idiopathic -
Benign childhood epilepsy with centro-temporal
spikes
- Symptomatic
- Temporal,
frontal, parietal, occipital lobe epilepsies
- Generalized
- Idiopathic
- Cryptogenic
or symptomatic - West, Lennox-Gastaut syndrome
OR
4 main types
|
Idiopathic Partial Epilepsy (20%) Mild,
focal onset seizures in children Usually
normal intellect Treatment
often minimal required e.g.
Rolandic epilepsy |
Symptomatic Partial Epilepsy (40%) Focal
onset seizures Lesions
of the cortex Usually
normal intellect Treatment
– medication and surgery - First line Carbamazepine e.g.
TLE, AD frontal lobe epilepsy |
|
Idiopathic Generalised Epilepsy (30%) Generalised
seizures Inherited
ion channelopathies Normal
intellect Treatment
– medication usually effective -
First line Valproate (e.g.
JME) |
Symptomatic (secondary) Generalised Epilepsy
(10%) Frequent,
severe, generalised seizures Intellectual
disability Treatment
– hard to control e.g.
Lennox-Gastaut Syndrome |
ILAE 2021
classification of epilepsy syndromes
Classification
First determine if
the epilepsy fits into a specific electroclinical syndrome or constellation
If
not classify on the presence or absence of a structural or metabolic condition
(presumed cause) and then on the basis of the primary mode of seizure onset
(generalized vs. focal)
Electroclinical syndromes by age
Neonatal and infancy
• Self-limited epilepsies
o Self-limited neonatal epilepsy
o Self-limited familial neonatal-infantile epilepsy
o Self limited infantile epilepsy
o GEFS+
o Myoclonic epilepsy in infancy
• Developmental and epileptic encephalopathies (DEE)
o Early infantile developmental and epileptic encephalopathy (EIDEE) (prev. Ohtahara syndrome + early myoclonic epilepsy)
o Epilepsy in infancy with migrating focal seizures (EIMFS)
o Infantile epileptic spasms syndrome (IESS) (Prev. West Syndrome)
o Dravet syndrome (DS)
• Aetiology specific syndromes
o GLUT1 DS
o Sturge-Weber syndrome
o Gelastic seizures with hypothalamic hamartoma
o Others…..
Childhood
• Self limited epilepsies of childhood
o Self-Limited Epilepsy with Centro-Temporal Spikes (SeLECTS) (Benign Rolandic epilepsy)
o Self-Limited Epilepsy with Autonomic Seizures (SeLEAS) (Panayiotopoulos syndrome, early onset benign occipital epilepsy)
o Childhood Occipital Visual Epilepsy (COVE) (Late onset benign occipital epilepsy, Gastaut-type)
o Photosensitive Occipital Lobe Epilepsy (POLE) (Idiopathic photosensitive occipital lobe epilepsy)
• Genetic generalised epilepsies
o Childhood absence epilepsy (CAE)
o Epilepsy with eyelid myoclonia (EEM) (Jeavons syndrome)
o Epilepsy with myoclonic absence (EMA) (Bureau and Tassinari syndrome)
• DEEs
o
Lennox-Gastaut
syndrome (LGS)
o Epilepsy with Myoclonic Atonic Seizures (EMAtS) (Doose syndrome)
o Developmental/Epileptic Encephalopathy with Spike Wave Activation in Sleep (DEE or EE -SWAS)
- (Including subtype Landau-Kleffner syndrome)
o Febrile Infection Related Epilepsy Syndrome (FIRES)
o Hemiconvulsion-heiplegia Epilepsy Syndrome (HHE)
Onset at variable age
• Generalised epilepsy syndromes
o Juvenile absence epilepsy (JAE)
o Juvenile myoclonic epilepsy (JME)
o Epilepsy with generalized tonic–clonic seizures alone (GTCA)
• Focal epilepsy syndromes (genetic)
o Self Limited (see childhood above)
- COVE
- POLE
o Familial mesial temporal lobe epilepsy (FMTLE)
o Epilepsy with auditory features (EAF)
o Sleep related hypermotor epilepsy (SHE) (Previously ADNFLE)
o Familal focal epilepsy with variable foci (FFEVF)
• Focal and generalised
o Epilepsy with reading induced seizures (EwRIS)
• Epilepsy with DE/EE
o FIRES (See above)
o Progressive myoclonic epilepsy
• Aetiology-specific
o Mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE-HS)
o Rasmussen Syndrome (RS)
Specific Epilepsy Syndromes
– Genetic Generalised Epilepsy
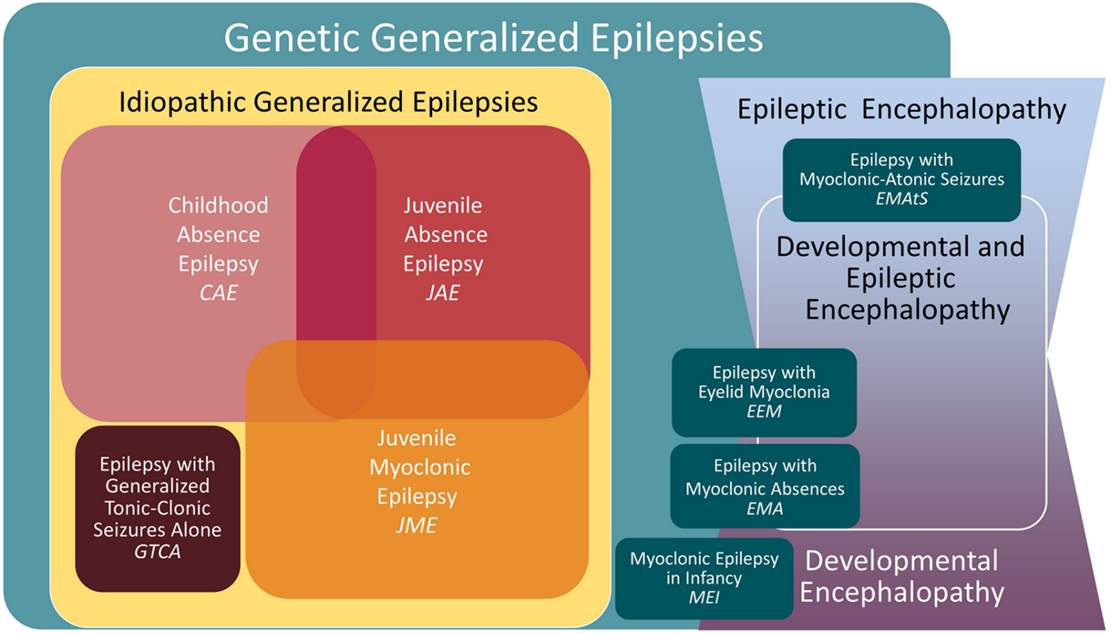
Idiopathic Generalised Epilepsies
Childhood Absence Epilepsy (CAE)
• 4-8 years
• Absence
• GTCS 40%
• Normal intellect
• Underlying genes unknown
EEG
- 3Hz Spike-Wave
- Polyspike can be present
- Frontal predominant F3, F4, Fz and F7, F8
- Exacerbated by hyperventilation
- 80% of children with active CAE will have seizures induced by HV
Treatment
• Ethosuximide (prevents absence but not GTCS)
• VPA
• LTG
Juvenile Absence Epilepsy (JAE)
• Onset adolescence
• Seizures
• GTCS
• Absence
• Less frequent absence seizures compared to CAE - ~1-2/week
• Each seizure is however longer ~20sec
EEG
• Similar to JME (faster than CAE)
Treatment
• VPA, LTG
•
Early onset absence epilepsy (not accepted as official category in ILAE)
• Onset <4years
• High frequency of developmental delay
• 10% due to GLUT1 deficiency
Juvenile Myoclonic Epilepsy (JME)
• Early
adolescence (onset from 10-16years)
• Polygenic cause
• 4% evolve from CAE
• Generalized seizures – absence seizures predominate at onset
• Bilateral myoclonic jerks – usually in the morning
o Consciousness usually preserved during jerks
o Single or repetitive
• Most frequent in the morning after waking, provoked by sleep deprivation
EEG
• Fast spike-wave discharges – 4-5Hz
• Polyspike
• Small percentage exhibit photoparoxysmal response
• Activated by hyperventilation and slow wave sleep
• Suppressed during REM sleep
• Epileptiform abnormality present in 90% on first EEG
Treatment
• Benign, responds well to treatment (valproate), usually does not remit (although may reduce in later life)
Epilepsy with tonic-clonic seizures alone (GTCA)
• GTCS with no other seizure types
• Onset 10-25yrs (range 5-40yrs)
• Seizure tendency usually lifelong and requires ongoing therapy
Genetic Epilepsy with febrile seizures
• Febrile seizures extending beyond 6 years +/- development of afebrile seizures
• Usually mild and resolves by adolescence
• Some due to Na+ channel mutations
• Some severe phenotypes
Other Genetic Generalised Epilepsies
Epilepsy with Myoclonic Absence (EMA)
• Onset 2-12
• EEG looks like CAE
• Jerking tonic seizures
• Absence seizures often atypical
• Can be intellectually normal or have features of DEE
Epilepsy with eyelid myoclonia (EEM) (Jeavons syndrome)
• Triad
o Frequent eyelid myoclonia
o +/- absence seizures
o Induced by eye closure and photic stimulation
• 1.2-2.7% of epilepsy
• Onset peak 6-8 (range 2-14)
• Female:male 2:1
• Usually normal intellect (some exceptions)
• Eyelid myoclonia can be associated with absence seizures with mildly impaired awareness
• Can also be absence seizures separate to the eyelid myoclonia
• GTCS occur in most patients, but are infrequent
• GTCS can often be controlled with ASMs, however eyelid myoclonia often not controlled
• Eyelid myoclonia may not be associated with EEG change in later life (i.e. a movement disorder)
•
GLUT 1 Deficiency (aetiological classification rather than true syndrome)
• Can have variable phenotypical presentation
• Can present as CAE phenotype
• Absence seizures
• Paroxysmal exercise induced dyskinesia (PED)
•
Lennox-Gastaut
Syndrome
• Defined by:
o Multiple/mixed seizure types
o EEG showing slow spike and wave
o Impaired cognitive function
Epidemiology and aetiology
• Children peak 2-5 years
• M >F
• Associated with a range of CNS diseases including: developmental abnormalities, trauma, infection, hypoxia etc.
• Can evolve out of a previously recognised encephalopathy/cerebral injury OR occur ‘de novo’
•
Clinical
• Seizure types
o Atypical absence
o Atonic seizures
o Tonic seizures
o Myoclonic seizures
• Poor prognosis
EEG
• Background
o Generalised and/or focal slow spike wave (1-2.5Hz) or polyspike
o Activated by sleep sometimes
o Brief bursts of rapid spikes in sleep
o Many patients manifest multifocal spikes or sharp waves
• Absence seizures
o Slow spike-wave – usually diffuse distribution
o Often less sharply demarcated onset and offset
• Atonic seizures
o
• Tonic seizures
o Brief runs of rapid spikes
Epilepsy with myoclonic atonic seizures (EMAtS) (Doose syndrome)
- Phenotype somewhat similar to LGS
- Myoclonic –astatic seizures more prominent
- Genetic (rather than lesional) cause
- Onset 1.5 to 5 years
- Variable response to treatment
EEG
- Background slow – 4-7Hz
- Slow spike wave during sleep – usually not as prominent as in LGS
- Myoclonic
Infantile epileptic spasms syndrome (IESS) (West Syndrome)
• Encompasses classic West Syndrome, but also those with spasms who do not meet all three criteria of Wests.
• West Syndrome - Triad of:
o Infantile spasms
o Hypsarrhythmia on EEG
o Neurodevelopmental disorder
• Onset between 3 months and 3 years
Aetiology
• Multiple possible underlying aetiologies (>200), often structural e.g.
o Tuberous sclerosis
o Cortical dysplasia
o Tumour
o Stroke, hypoxic ischaemic encephalopathy
o Genetic – Trisomy 21, many single gene disorders
• Aetiology unknown in 30%
Clinical
• Infantile spasms are the hallmark
o Flexion, extension or mixed type
o Flexor spasms
- Brief tonic contraction in flexion at hips and neck
- Tensing of the shoulders – sometimes in abduction
- “Jack-knife seizures”
o Typically held for 1 sec (i.e. longer than a myoclonic seizure)
o Tend to occur in clusters that may last for several minutes
• A proportion will go on to develop LGS
EEG
• Hypsarrhythmia (Greek – ‘high’ or ‘lofty’)
• High voltage, chaotic rhythm
• Activated by sleep
o Suppressed in REM sleep
• Modified Hypsarrhythmia
o When does not quite meet full definition
o One example would be when activity is only seen during sleep and the awake EEG is relatively normal
• Spasms
o Complex slow wave followed by low voltage, rapid spikes
Management
• Depends on underlying cause
• ACTH
• Corticosteroids
• Vigabatrin
(Particularly effective if tuberous sclerosis is underlying cause)
• Ketogenic diet
• Levetiracetam
• Benzo’s
• VPA
• Surgery
Dravet Syndrome
• Previously known as Severe Myoclonic Epilepsy of Infancy
Clinical
• Normal infant
• Seizures from 6 months
• Focal clonic/Hemiclonic or tonic-clonic seizures
• Often associated with trigger:
o Fever (can initially appear like a typical ‘benign’ febrile seizure)
o Infection
o Environmental heat/hot baths
o Immunisation
o Sunlight
o Pattern stimulation
o Exercise
o Excitment
• Prolonged seizures/status epilepticus then develop
• Frequent seizures over next 6/12
• Other seizures by 1-4yrs
o Myoclonus
o Absence
o Focal seizures
o Tonic
o Atonic
• Other features
o Developmental delay
o Crouching gait
o Speech deficits almost universal
o Parkinsonism can develop in later life
EEG
• Initial interictal EEG is often normal
• No characteristic features
Aetiology
• 80% have SCN1A mutation (sodium channel)
• 95% de-novo mutation
• Type of mutation can correlate with severity of phenotype
o Truncating mutations generally worse
• Can overlap with GEFS+ (which is also due to SCN1A mutation)
Treatment
• Avoid triggers
o Aggressively treat fevers and infections
• Avoid sodium channel blockers (given SCN1A encodes a Na Channel)
o It is unclear if this is absolute in adult patients. Some adult patients have been observed to have a good response to lamotrigine.
o Phenytoin should be avoided as a long term treatment but may be effective in status epilepticus

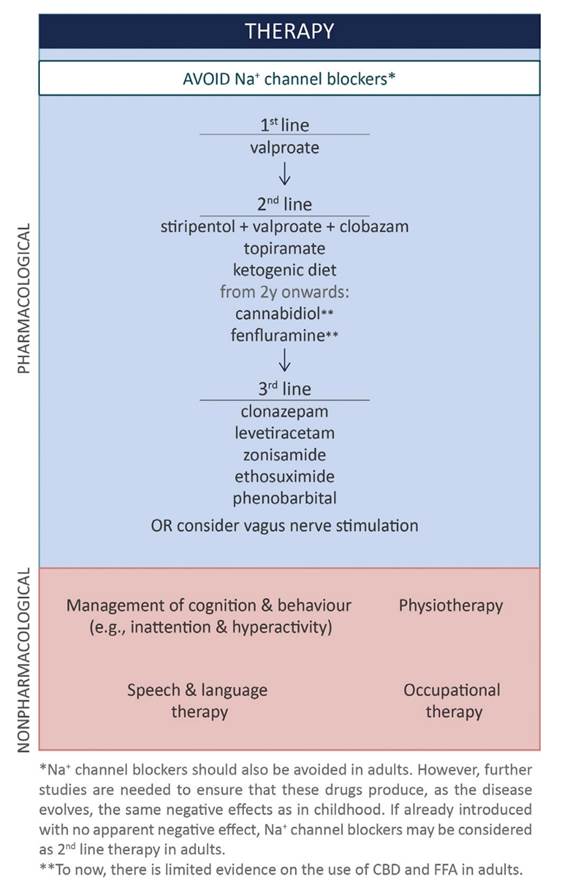
• Stiripentol
o Reduces seizure frequency (13% in one study c.f. 51% with valproate)
o May be particularly effective at decreasing length/severity of status epilepticus
o Probably less useful in older patients
• Canabidiol
o RCT demonstrated 41% reduction vs 16% in placebo group
o Can push up clobazam levels
• Fenfluramine
o Old serotonergic drug, previously used as anti-obesity, was associated with valve disease in old trials
o 50-70% response rate vs ~7% in placebo
o Well tolerated in trials – no evidence of valve disease
• Ketogenic diet
o Some evidence of efficacy
o Less tolerated as patient ages
• Behaviour management
o Methylphenidate used in some cases
Prognosis
• 15-20% mortality
• Largely due to SUDEP
• Developmental delay
References:
• Guidance on Dravet Syndrome, Epilepsia open 2021
Progressive Myoclonic Epilepsies
• Myoclonic
• GTCS
• Progressive neurological deficits
• Causes
o Unverricht-Lundborg disease
o MERFF
o Lafora
o Neuronal ceroid lipofuscinosis
Progressive, dementing conditions
Neuronal ceroid lipofuscinosis
•
Lafora body disease
• Mutation in Laforin gene
• Diagnosis by detection of Lafora bodies in skin biopsies
• Onset 6-19
• GTCS and myoclonic seizures
• Also focal seizures with preserved awareness and visual seizures
• Condition progresses with severe ataxia, myoclonus and dementia
• Death occurs 2-10 years after onset
Non-dementing
MERRF
• See Mitochondrial disease topic
• Variable age of onset
Unverricht-Lundborg disease (Baltic Myoclonus)
• Cystatin B mutation
• Onset age 7-16
• Action myoclonus
• Later GTCS
• Ataxia
• Can be worsened by phenytoin
MEAK
• Myoclonic and Epilepsy with Ataxia and Potassium Chanel
• Described by Austin group (2015)
• Onset 5-15
• Looks like JME to start with but then progresses to refractory myoclonus
• Wheel-chair bound in late teens due to myoclonus
• Cognition generally normal
• De novo mutation in potassium channel
Familial Adult Myoclonic Epilepsy (FAME)
· Tremor and seizures (?focal or generlaised
· EEG – not always abnormal – usu generlaised
Developmental Epileptic Encephalopathy – Spike and wave activation in sleep (DEE-SWAS/EE-SWAS) – including Landau-Kleffner
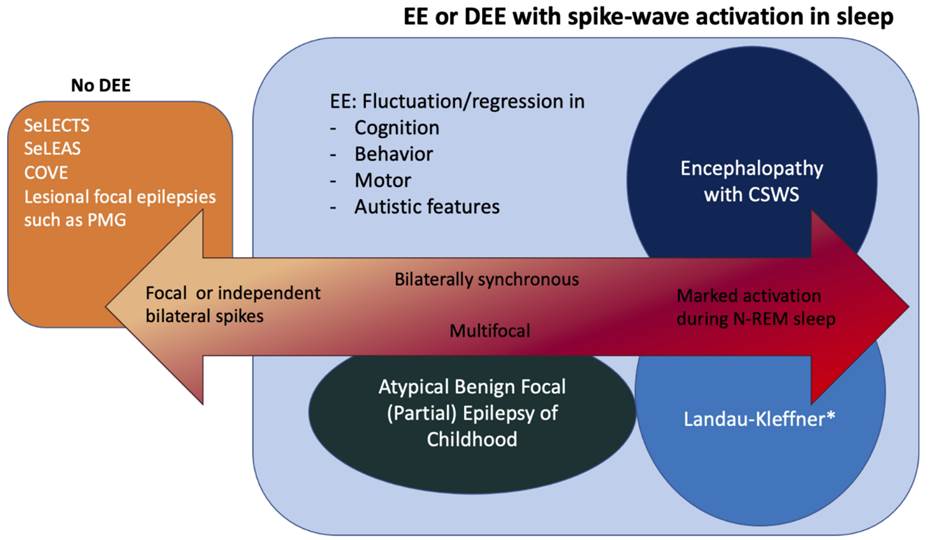
• Normal development with isolated language delay
• Speech regression with auditory agnosia
• Behavioural difficulties
• 70% mild epilepsy
EEG
• Continuous spike-wave in sleep (CSWS) for 85% of non-REM sleep
Concept of Developmental/Epileptic Encephalopathies
· The pattern in which brain reacts to an insult depends on the patients age
· Thus the concept of age- dependent epileptic encephalopathies
· The three main patterns are (all described in detail above):
o EIEE
o WEST Syndrome
o LGS
Genetic focal epilepsies
Sleep related hypermotor epilepsy (SHE)
• Previously ADFLE
Familial Mesial Temporal Lobe Epilepsy (FMTLE)
Familial Focal Epilepsy with Variable Foci (FFEVF)
Epilepsy with Auditory Features (EAF)
Self-Limited Focal Epilepsies (SeLFEs)
Childhood occipital visual epilepsy (COVE)
- Previously called: Late onset benign occipital epilepsy, Gastaut syndrome, idiopathic childhood occipital epilepsy-Gastaut type
Photosensitive occipital lobe epilepsy (POLE)
- Rare 0.7% of childhood epilepsies
Idiopathic Focal Epilepsies
Self-Limited Epilepsy with Centrotemporal Spikes (SeLECTS)
(Benign Rolandic Epilepsy)
·
10-20% of childhood epilepsies
·
Onset 3-13yrs
·
Most common – simple partial seizure involving the
face
·
Originate from perisylvian
(rolandic) sensorimotor cortex which represents face
and oropharynx
·
Motor activity in upper extremity can also occur
·
¾ seizures occur at night or on awakening
·
50% have a tonic-clonic
seizure at some time.
·
EEG – Centrotemporal sharp waves with distinctive
features
o
Biphasic, negative sharp peak followed by a
positive rounded component
o
Occur in 1-2% of asymptomatic children as well
·
Spontaneous remission in majority by age 13yrs
Self-Limited Epilepsy with Autonomic Seizures (SeLAS)
(Panayiotopoulos syndrome)
Specific epilepsy
syndromes – “Symptomatic/Lesional” Focal Epilepsies
Mesial
Temporal Lobe Epilepsy with hippocampal sc
- Commonest
cause of adult epilepsy
- Frequently a
history of early cerebral insult
(febrile seizure
- Complex
partial seizures
- Characteristic
hippocampal sclerosis
- Auras common
- Visceral(epigastric),
gustatory, dysmnestic, affective
- Partial awareness
commonly preserved
- Dystonic
posturing of the contralateral upper limb
- Infrequent
secondary generalized seizures
- Prominent
motor arrest – motionless stare
- Refractory to
medication, responds well to surgery
Hypothalamic
Hamartoma
• Precocious puberty
• Laughing seizures from infancy
• Evolve into partial seizures
Focal Cortical Dysplasia
• Blumcke Classification (2011)
|
Type |
|
|
|
|
I |
Focal cortical
dysplasia with abnormal cortical lamination |
|
|
|
II (Taylor type) |
Focal cortical dysplasia
with dysmorphic neurons |
A: Without balloon
cells B: with balloon
cells |
Radial-glial bands
on MRI |
|
|
|
|
|
|
III |
Architectural
distortion of the cortical layer |
A: in temporal lobe
with hippocampal atrophy B: adjacent to glial
or glioneuronal tumour C: Adjacent to
vascular malformation D: Adjacent to
other lesions acquired in early childhood |
|
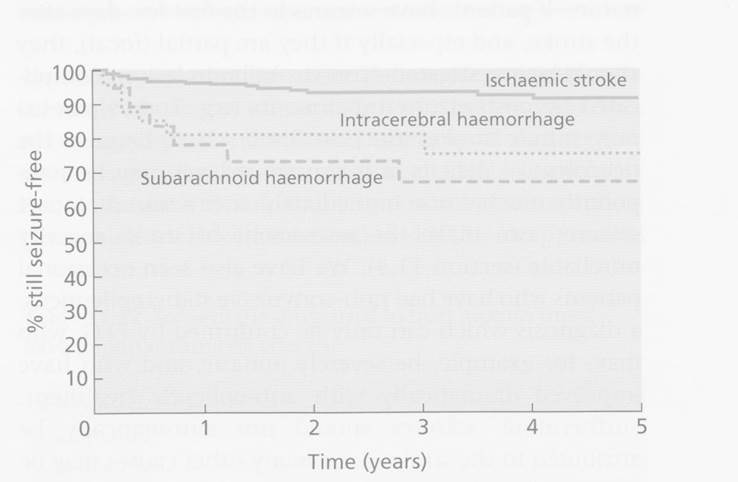
Post Stroke Seizures
·
5% of patients will have a seizure within first week (most in first
24hours)
o
More common in haemorrhagic and strokes affecting cortex
·
Later seizure risk
o
3-5% in first year and 1-2%/year thereafter
o
3% of all patients have >1 seizure and could be considered epileptic
o
This represents 20x increased risk compared to control population.
o
Risk of recurrent seizure after first seizure is 50%.
|
Gene |
Function |
|
Clinical Syndromes |
Benefit of testing |
|
SCN1A |
Sodium channel type 1alpha |
|
Dravet syndrome OR Genetic Epilepsy with Febrile
Seizures Plus (GEFS) OR Early infantile SCN1A encephalopathy
(rare) |
Diagnosis AED choice (sodium channel blockers
make things worse – except lamotrigine).
VPA and Benzodiazepines good. Stiripentol
useful. |
|
SLC2A1 |
Mutations in GLUT1 |
Usually de novo mutations |
GLUT1 Encephalopathy Early-onset childhood absence
epilepsy Overlap with movement disorders |
Diagnosis Ketogenic diet |
|
PCDH19 |
Protocadherin 19 |
|
Female patients with clustered focal
seizures, (onset under age 3, often with intellectual disability. |
Counselling |
|
KCNQ2 |
|
|
Epileptic encephalopathy OR Benign epilepsy syndrome |
?Ezogabine in future |
|
KCNT1 |
Sodium activated potassium channel |
|
Autosomal dominant frontal lobe
epilepsy (ADNFLE often with psychiatric features and ID) OR Epilepsy in infants with migrating
seizure focus (EIMFS) – poor prognosis |
?Quinidine |
|
?CHRN |
|
|
ADNFLE |
|
|
PRRT2 |
Binds to SNAP25 - |
|
Paroxysmal Kinesogenic
Dyskinesia (PKD) - overlaps with Benign familial infantile epilepsy |
Carbamazepine or oxcarbazepine might
be effective |
|
TSC1 and TSC2 |
|
|
Tuberous sclerosis |
Vigabatrin might be effective ?Rapamycin might be effective |
|
ALDH7A1 and PNPO |
|
|
Severe, early onset epilepsy |
Pyridoxine |
|
Experimental |
|
|
|
|
|
DEPDC5 |
DEP containing domain 5 |
|
Familial focal epilepsy with variable
foci |
|
|
GRIN2A |
NMDA receptor subunit epsilon1 |
|
Landau-Kleffner
syndrome Epilepsy aphasia disorders |
|
|
|
|
|
|
|
Familial Focal Epilepsy
• First degree relatives of focal epilepsy patients have a 2.5x risk of epilepsy
Autosomal Dominant Sleep-related Hypermotor Epilepsy (ADSHE)
• Previously Autosomal dominant frontal lobe epilepsy (ADFLE)
• Onset in childhood
• Seizures
o Brief tonic or hyperkinetic
o Tend to occur in clusters
o Occur during sleep – shortly after falling asleep or before waking
o Aura’s common
o Often with preserved awareness during seizures
• Treatment
o Carbamazepine
• Prognosis
o Usually relatively benign
• Aetiology
o CHRN Mutations (Nicotinic AChR)
o DEPDC5
o KCNT1
o Mutation has some correlation with severity of phenotype
Autosomal Dominant Epilepsy with Auditory Features
• Seizures that originate in the lateral temporal lobe
• Seizures:
o Prominent auditory aura
o Sometimes receptive aphasia
o Usually poorly formed sounds (ringing, buzzing)
o Sometimes voices
o Sometimes a sudden loss of sound
o Can have focal aware, FIAS and FBLTC
• Aetiology:
o LGI1 in 30-50%
o RELN mutations (17%)
Familial Focal Epilepsy with Variable Foci (FFEVF)
• Focal epilepsy has variable foci in different family members, but constant focus within individual
o E.g. Families with temoral, frontal, parietal and occipital lobe epilepsies
• Variable age of onset and severity
• May be co-morbid psychiatric conditions
• Often lesion negative, however some cases associated with focal cortical dysplasia and subcortical grey matter heterotopia
• Aetiology:
o DEPDC5 ~80% of families
- Protein is a member of the GATOR1 complex which inhibits the mTOR pathway
o NPRL2 and NPRL3 genes (encode different parts of GATOR1 pathway)
Familial mesial temporal lobe epilepsy
• Defined by mesial temporal lobe epilepsy with strong family history (otherwise can look like sporadic MTLE)
• Onset adolescence or early adulthood
• Seizures:
o Focal aware seizures with prominent psychic or autonomic features
o Intense déjà vu, dream like states, flashbacks, fear, epigastric sensations, flushing, olfactory sensations
o Sometimes FIAS
o Infrequent FBTC (often from sleep)
• Aetiology
o Most cases unknown
o DEPDC5 in a small percentage of cases
o ~20% of cases of MTLE
• Relatively benign and responsive to medication compared to other forms of MTLE
Other:
• Familial neonatal epilepsy
• Familial neonatal-infantile epilepsy
• Familial infantile epilepsy